Inheritance and manifestations of albinism
Albinism is a kind of hereditary leukoplakia caused by melanin deficiency or synthesis disorder of skin and accessory organs due to tyrosinase deficiency or functional decline.
According to clinical manifestations, albinism can be divided into three categories: ocular albinism (OA), ocular skin albinism (OCA) and albinism-related syndrome, among which ocular skin albinism (OCA) is the most common and albinism-related syndrome is the rarest.
This paper mainly expounds the clinical manifestations, genetic causes and genetic counseling of OA and OCA.
Clinical manifestations of OA and OCA
Only the eye pigment in OA patients is reduced or lacking, while the eye, skin and hair in OCA patients are obviously reduced or lacking.
1, eye performance
① Iris transillumination defect (TID)-91% ~ 100% albino patients have TID.
② nystagmus
③ Fundus hypopigmentation —— It is caused by the decrease of retinal pigment epithelium and/or choroidal pigmentation, which often leads to obvious choroidal vessels in the posterior pole. It is reported that more than 94% patients with albinism have fundus hypopigmentation.
④ hypoplasia of fovea-characterized by the continuation of the inner layer of retina behind fovea and the decrease of cone photoreceptor specialization.
⑤ optic nerve abnormality
⑥ ametropia
2. Skin manifestations
The skin and hair color of albino patients may change with age.
① Skin and hair pigmentation score
Skin and hair pigmentation scores help to objectively determine the influence of hypopigmentation.
② Skin diseases
Including all albinos, regardless of skin color, the risk of ultraviolet (UV) light-related skin diseases will increase. The most important of these is skin cancer, which includes squamous cell carcinoma, basal cell carcinoma, melanoma and less common Merkel cell carcinoma.
Genetic causes of OA and OCA
The following table summarizes the genes known to be related to nonsyndromic OCA and OA. All nonsyndromic OCA are inherited by autosomal recessive mode. OA (always non-syndrome) is inherited in an x-linked manner.
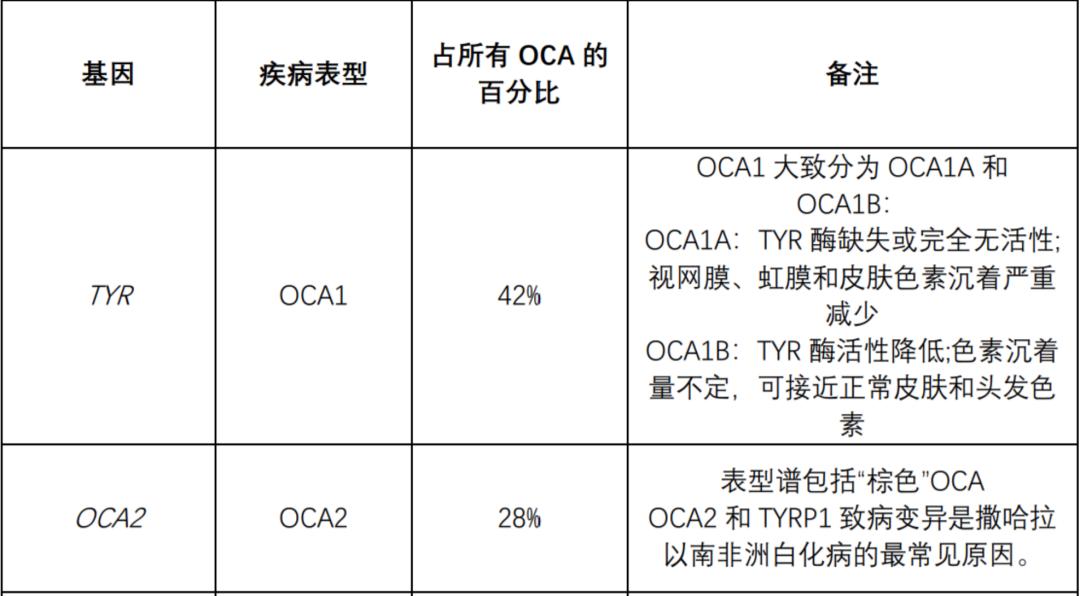
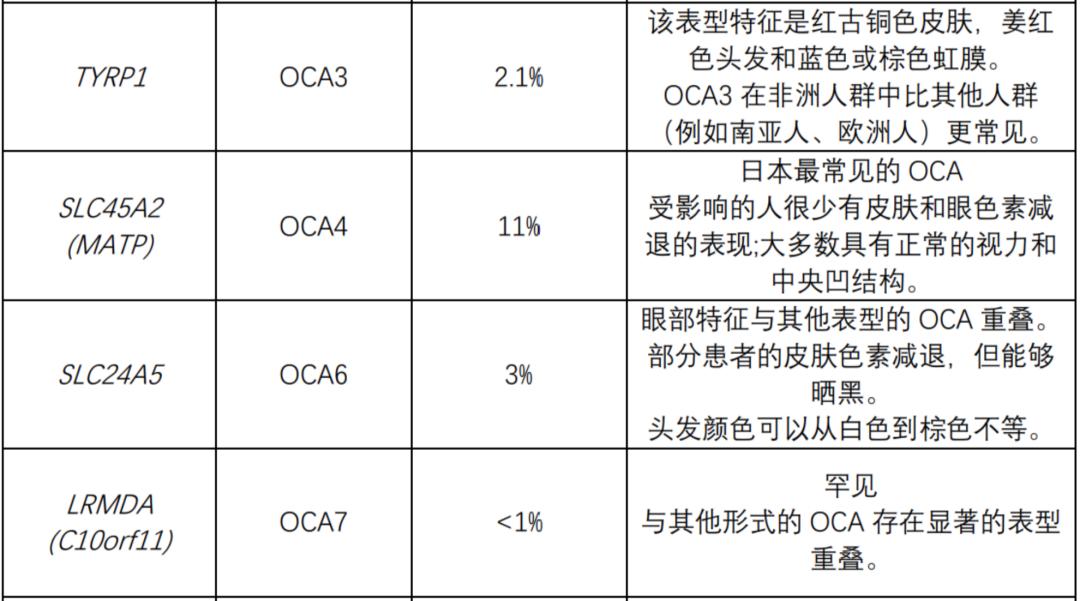
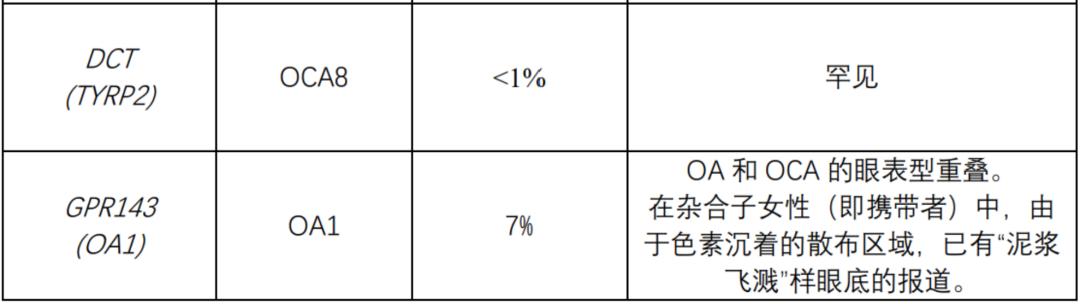
The following table summarizes the genes known to be related to OCA syndrome:
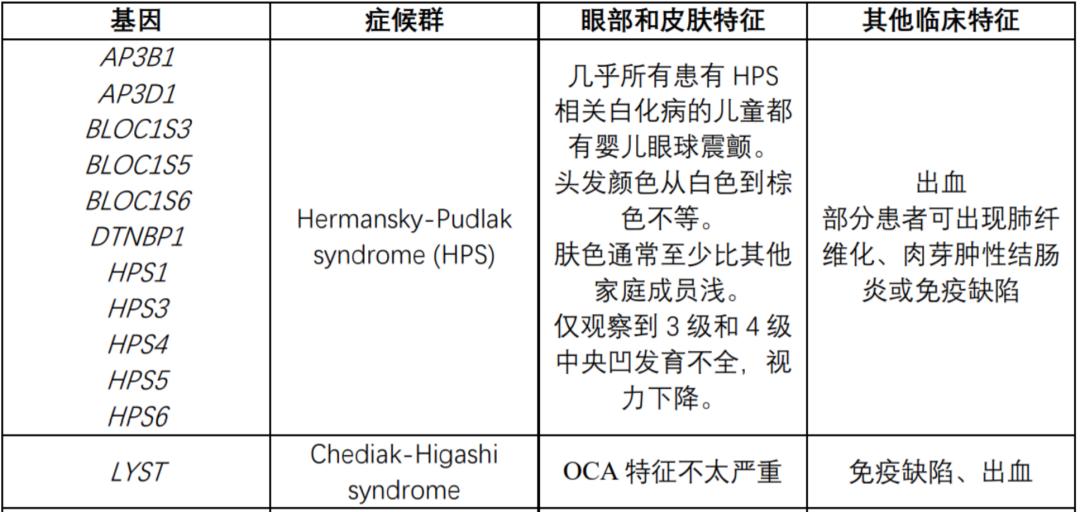

Genetic counseling
1. Inheritance mode
Nonsyndromic ocular skin albinism (OCA) caused by pathogenic variation in TYR, OCA2, SLC45A2, SLC24A5, LRMDA (C10orf11) or DCT (TYRP1) is inherited in autosomal recessive way.
Ocular albinism (OA) caused by pathogenic variation in GPR143(OA1) is inherited by X linkage.
If an individual suffers from a specific syndrome related to OCA (such as Hermansky-Pudlak syndrome or Chediak-Higashi syndrome), genetic counseling is needed for this disease.
2. Autosomal recessive inheritance-risk to family members
Parents of the proband:
The parents of the affected children are presumed to be heterozygotes causing the pathogenic variation of OCA.
If molecular diagnosis has been established in the proband, it is suggested that the parents of the proband should be tested for molecular genes to confirm that both parents are heterozygotes causing the pathogenic variation of OCA and allow reliable recurrence risk assessment.
If the pathogenic variation is detected only in one parent, and the parental identity test has confirmed biological motherhood and parent-child relationship, one of the pathogenic variations identified in the proband may occur as a newborn event in the proband or as a zygote in the mosaic parent. If the proband seems to have homozygous pathogenic variation (that is, the same two pathogenic variations), other possibilities to be considered include:
The deletion of single exon or multiple exons in the proband was not detected by sequence analysis, which led to artificial homozygosity;
Parental chromosomes and single parent homomorphism of pathogenic variation lead to homozygosity of pathogenic variation among proband.
Heterozygotes (carriers) are asymptomatic and have no risk of developing the disease.
Siblings of the proband:
If it is known that both parents are heterozygotes of pathogenic variants that cause OCA, each sibling of the affected individual has a 25% chance of being affected at conception, a 50% chance of becoming asymptomatic carriers, and a 25% chance of inheriting any familial pathogenic variants.
Heterozygotes (carriers) are asymptomatic and have no risk of developing the disease.
Descendants of the proband:
The offspring of autosomal recessive OCA individuals are obligate heterozygotes (carriers) that cause the pathogenic variation of OCA.
Other family members:
Every brother and sister of the proband’s parents has a 50% risk of becoming the carrier of OCA’s pathogenic variation.
Carrier detection:
It is necessary to identify the pathogenic variation that causes OCA in the family in advance for carrier detection of at-risk relatives.
3. X-linked inheritance-risk to family members
Parents of male proband:
The father of the affected man will not suffer from this disease, nor will he be a hemizygote of the pathogenic variant GPR143; Therefore, he does not need further evaluation/testing.
In a family with more than one affected individual, the mother of the affected male is an obligate heterozygote. Note: If a woman has more than one affected child and no other affected relatives, and if family pathogenic variation cannot be detected in her white blood cell DNA, she is likely to have germline chimera.
If the male is the only affected family member (i.e., a simple case), the mother may be heterozygote, the affected male may have new pathogenic variation (in this case, the mother is not heterozygote), or the mother may have somatic/germline chimera.
It is suggested that the mother should be tested for molecular genes to confirm her genetic status and make a reliable recurrence risk assessment.
Brothers and sisters of male proband:
The risk of siblings depends on the mother’s genetic status:
If the proband’s mother has the pathogenic variation of GPR143, the chance of transmission in each pregnancy is 50%.
Men with genetic disease variation will be affected;
The female with genetic pathogenic variation will be heterozygote (i.e. carrier). Heterozygotes are usually unaffected, although they can show iris penetration defects, "mud splash" fundus appearance and fovea hypoplasia.
If the proband represents a simplex case and the pathogenic variation cannot be detected in the mother’s leukocyte DNA, it is assumed that the risk of sibs is low, but the risk is higher than that of the general population because of the possibility of maternal lineage mosaic.
Descendants of male proband:
The affected men spread the disease-causing mutation of GPR143 to all their daughters, but not to their sons.
Other family members:
Menstruation and maternal cousins of the male proband may be at risk of GPR143.
Note: Molecular genetic testing may be able to identify family members with new pathogenic mutations, and this information can help determine the genetic risk status of large families.
Heterozygote detection:
To identify female heterozygotes, it is necessary to identify the pathogenic variation of GPR143 in the family in advance.
Note: As carriers of this X-linked disease, women are heterozygotes, which are usually not affected, but they may show iris penetration defects, "mud splash" fundus appearance and fovea hypoplasia.
Source | Metz Medical official website
Write | |Anna.An
Original title: Inheritance and Manifestation of Albinism
Read the original text